

Traditionally, peanut cultivar development has been dominated by conventional breeding methods. These efforts have resulted in increased yield (Kanuft and Wynne, 1995; Isleib et al., 2001) and disease resistance (Simpson and Starr, 2001; Holbrook et al., 2008) as well as improvements to peanut seed composition like high oleic oil (Knauft et al., 1993; Isleib et al., 1996). However, these methods are limited in that they are dependent on measurable phenotypic characteristics such as morphology and chemical traits. Marker-assisted selection (MAS) has been integrated into peanut breeding practices in recent years (Guo et al., 2011). This process utilizes genetic markers associated with individual genes or with quantitative trait loci (QTLs) to better measure the possible phenotypic variations within individuals allowing for increased efficiency in breeding selection resulting in higher rates of genetic gain. In order to generate more detailed genomic information for MAS, several marker-based methods have been applied such as the use of microsatellite or SSR markers for genetic maps (Varshney et al., 2009; Hong et al., 2010).
SSRs (simple sequence repeats) or microsatellite markers or sometimes referred to as a variable number of simple repeats are short segments of DNA that have a repeated sequence. The repeated unit may occur different times in different lines in the same crop species or population. Therefore, SSRs are a class of molecular markers based on polymerase chain reaction (PCR) primers designed to flank a set of tandem repeats of a short DNA sequence. Insertion or deletion of these repeat units contributes to the marker polymorphism (Liang et al., 2009; Wang et al., 2009). SSRs are highly polymorphic, multi-allelic, co-dominant, ubiquitously distributed in the genome, analytically simple, and readily transferable between species such as from soybean to peanut (He et al., 2006), and have been applied in recent years for the construction of genomic linkage maps and QTL analyses of various plant species including peanuts (Varshney et al., 2009; Hong et al., 2010; Qin et al., 2011). SSR marker polymorphism due to insertion or deletion of a short tandem repeat can be detected by different sizes of PCR amplicons. These amplicons can then be visualized using gel electrophoresis to determine the length of the products. Several methods have been proposed for the electrophoresis of these PCR products. In instances where size difference between fragments are large, standard high-melting agarose or MetaPhor agarose gels stained with ethidium bromide or SYBR green can be utilized. However, in most cases, size difference between fragments are relatively small (2–20 bp) and require the use of methods producing fine resolution such as polyacrylamide gel electrophoresis (PAGE) in conjunction with visualization methods such as fluorescent dye labeling or silver staining. Of these methods, silver staining is relatively simple, low cost, and more sensitive to the detection of nucleic acid fragments of low concentration in polyacrylamide gels than intercalating fluorescent dyes such as ethidium bromide and SYBR green (Rahman et al., 2000, Hwang et al., 2006).
In the past, given the cost involved in PCR amplification and gel electrophoresis of the large numbers of markers required in genotyping studies, laboratories that possess a small budget and/or limited or no access to expensive equipments are severely hindered to conduct this type of research activities. In this study, we intended to introduce and describe a method that is easy to handle, low-cost, and relatively high-throughput system for genotyping applications in peanut research.
Materials and Methods
Description of the electrophoresis apparatus
The electrophoresis system utilized in this method is the DYCZ-30B Electrophoresis Cell manufactured by the Beijing WoDeLife Sciences Instrument Company (http://www.ly.com.cn/, Beijing, China). The unit consists of two clamp-plates and allows simultaneous running of two gels. The two gels are cast between glass plates with dimensions of 20 cm wide and 10.5 cm high for the front plate, and 20 cm wide and 11.3 cm high for the rear plate. The rear plate also has built-in spacers measuring 1.0 mm in thickness. Upon assembly, final cast gel dimensions are 10.5 cm × 18.5 cm × 0.1 cm (H × W × T).
The sample capacity of each gel is dependent on the comb selected. The manufacturer offers various sized combs allowing for 25, 40, and 52 samples per gel. These configurations allow for the use of standard single-, eight- or 12-channel pipettes for loading although samples need to be carefully reordered in an alternating sequence when using multichannel pipettes in the 52 sample configuration. Using the 52 sample configuration, an assembled gel apparatus has a total capacity of 104 samples. In our applications with the 52 samples per gel configuration, 96 samples are loaded into a single assembled apparatus, which matches the 96-sample format of a PCR plate, and also allows for space on either side of the gels preventing errors due to the ‘edge effect’ observed during electrophoresis.
Electrophoresis protocol
Nondenaturing polyacrylamide gel is cast in a glass plate sandwich, in which the plates are first cleaned with 70% (w/v) ethanol, and then assembled with a gasket provided by the manufacturer. This gasket has slots to help position the glass plates correctly for the specified gel dimensions described in the previous section. The position of the front glass plate creates a tight seal with the gasket while the rear plate is positioned with a gap between the plate and the lower surface of the gasket. Once the glass plates are inserted into the gasket, this gap is sealed by pipetting 2% (w/v) molten agar. The molten agar is allowed to polymerize for approximately 1 minute. This is repeated twice for each glass plate sandwich used. Following polymerization of the agar seal, the two assembled plate sandwiches are placed into the rig so that the agar seal sides of the sandwiches are facing each other. Orientation of the plates is an important consideration since an electrode runs along the bottom-center of the rig and is shared by both plate sandwiches. The rig also has two additional electrodes, one at the top of each side plate.
Following assembly of the apparatus, an acrylamide gel solution is poured between the plates. Approximately 20 ml of gel solution is required for each gel. The gel consists of a final concentration of 6% (w/v) acrylamide/bis-acrylamide (29∶1) for isolation of large sized DNA fragments (≥ 500bp) or 9% (w/v) acrylamide/bis-acrylamide (29∶1) for isolation of small to medium sized DNA fragments (< 500bp), 0.5 X TBE buffer, 0.1% (w/v) ammonium persulfate, and 0.075% (w/v) N,N,N′,N′-tetramethylethylenediamine (TEMED). Following the addition of ammonium persulfate and TEMED, the gel solution is thoroughly mixed and immediately poured between the glass plates in the apparatus. A 52-tooth comb is then placed between the plates and the gel is then allowed to polymerize for approximately 10 minutes. Following polymerization, the electrophoresis apparatus tanks are filled with approximately 2.1 L of 0.5 X TBE buffer. The combs remain in place until immediately prior to loading of the samples. The gel can be kept in this manner for up to 24 hours.
The combs are then removed and DNA samples are loaded using a standard single-, eight- or 12-channel pipette. Two loading buffers are present in the DNA samples used in this study, the loading buffer component of the 5X Green GoTaq Reaction Buffer (Promega, Madison, WI, USA) used in the PCR reaction, and an additional Type III gel-loading buffer consisting of 0.025% bromophenol blue, 0.025% xylene cyanole FF, and 30% glycerol in water. A loading volume of 1.5µl is used in this method. After sample loading, electrophoresis is performed at voltage and timing dependent on the concentration of the gel used. For 6% polyacrylamide gels, electrophoresis is performed at 160 V for 1 h 20 min while electrophoresis of 9% gels is performed at 180 V for 1h 40 min.
Following electrophoresis, the gels are subjected to silver staining as described by Zhang et al. (2000) in order to visualize the resolved PCR products. In this process, the nondenaturing polyacrylamide gels are first removed from the plate sandwiches. If multiple gels are stained simultaneously, each may be labeled with holes punched in areas of the gel not expected to contain sample data. Removed gels are then placed in a large container with 1.0 L of fix buffer consisting of 10.0% (w/v) ethanol and approximately 0.5% (w/v) glacial acetic acid. The gels are incubated in fix buffer for a minimum of 5 min, but may be incubated as long as 1 hr. The fix buffer is then poured off and saved for use later in the procedure. The gels are then rinsed with deionized water. Following rinsing, 1.0 L of a 0.2% (w/v) AgNO3 staining solution is added to the container with the gels and incubated for exactly 12 min. Following staining, the gels are subsequently de-stained for 45 sec to 1 min depending on the number of gels utilized in the protocol in deionized water. Immediately after de-staining, the deionized water is poured off and 1.0 L of developer solution consisting of 1.5% (w/v) NaOH and approximately 5 mL of a 37% (w/v) formaldehyde solution is added. The gels are then incubated in the developer solution for 6 to 8 min.
After development, the gels are rinsed with deionized or tap water and placed back into the saved fix buffer. The gels may be stored in this fashion for up to 24 hrs until examined for data acquisition. The gels can then be placed on a white light box for visualization. The gels can also be covered with clear plastic wrap and the data points can then be scored based on the observed genotype. Gels were photographed in this study using a Cyber-shot DSC-H10 digital camera (Sony, Tokyo, Japan).
SSR markers and PCR protocol
Peanut SSR markers used in this study have been applied in genotyping of two mapping populations (Qin et al., 2011). The PCR amplification performed in this study is done utilizing both a PTC-225 DNA Engine Tetrad Peltier Thermal Cycler (MJ Research, Waltham, MA, USA) and a DNA Engine Tetrad 2 Peltier Thermal Cycler (BioRad Laboratories, Hercules, CA, USA). In addition, supplementary PCR amplifications are also performed using smaller units including a PTC-200 DNA Engine Peltier Thermal Cycler (MJ Research) and a GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA, USA). The PCR reaction volume is 15 µL and each reaction mixture contained 50 ng of template DNA, 0.5 µM of each primer (forward and reverse), 0.2 µM of each dNTP, 1.5 mM MgCl2, 1.0 unit of Taq polymerase, and 1 X Green GoTaq Reaction Buffer (Promega). The PCR is performed using a program as follows: initial denaturation at 95°C for 2 min, followed by 35 cycles of denaturation at 94°C for 45 sec, annealing at 51 – 55°C for 45 sec, and extension at 72°C for 1 min. A final extension at 72°C for 5 min is included at the end of the cycle. The PCR product is then stored at 4°C until analysis with PAGE.
Results and Discussion
The low-cost, high-throughput nondenaturing PAGE system described in this study was able to resolve alleles of SSR markers with varying length and degrees of polymorphism. This system was capable of separating alleles with size differences as little as 2 – 3 base pairs (bp); however, the system was found to be somewhat limited in its ability to observe such differences in low size range alleles (< 100 bp) because of the short length of the gel (10.5 cm) and short running time and lower power voltages (80 min to 100 min at 160 V to 180) in comparison with the large gel system and high voltage (22 cm and 350 V) (Wang et al., 2003). But this system described here is much easier to handle because of the smaller gel size.
For SSR markers with alleles greater than 200 bp in size, the system was found to be able to readily resolve differences in size between fragments. For medium size range alleles (200 – 500 bp), such as those of the SSR markers ARS213 and ARS286 with 2 or more alleles (Figure 1), the system was able to produce clear separation of all desired fragments as well as to discern polymorphisms of varying sizes. Low size range alleles were preferably resolved using a 9% acrylamide/bis-acrylamide gel; although a 6% (w/v) acrylamide/bis-acrylamide (29∶1) gel may be used for low size range alleles. The percentage of gel concentrations along with the voltages and running times are adjustable according to the markers and the polymorphic banding patterns and sizes. We used a 6% acrylamide/bis-acrylamide gel for most medium size range and all larger size range alleles (≥ 500 bp) in the genotyping study (Qin et al., 2011), such as those for the SSR marker ARS398 (Figure 2) with electrophoresis at 160 V for 1 h 20 min. In instances with medium and large size alleles with only slight differences in size, longer run times were used in order to obtain better resolution.
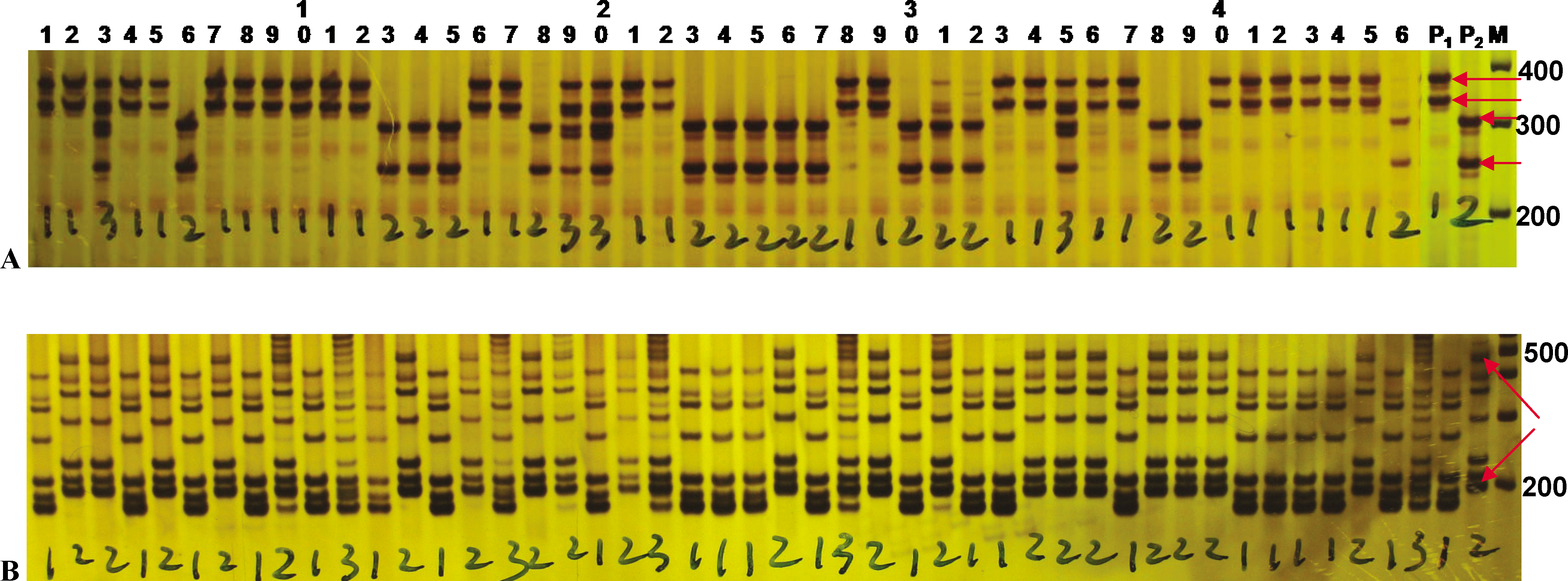
Polyacrylamide gel for the SSR marker ARS213 (A) and ARS286 (B) possessing 2 or multiple co-segregating alleles between 200 – 500 bp in length. These images were acquired following electrophoresis with a 6% (w/v) acrylamide/bis-acrylamide (29∶1) gel at 160 V for 1 h 20 min. The DNA fragments were visualized using silver staining. The polymorphic, co-segregating alleles are indicated by horizontal arrows. Hand-marked numbers visible on the image are scores representing the zygosity of the individual with “1” representing homozygous alleles for P1, “2” representing homozygous alleles for P2, and “3” representing a heterozygous combination of alleles from both P1 and P2. Lanes 1 – 46 are progeny of a segregating population at F2∶5 generation. P1 and P2 are two parents. M is marker of 100 bp DNA ladder.
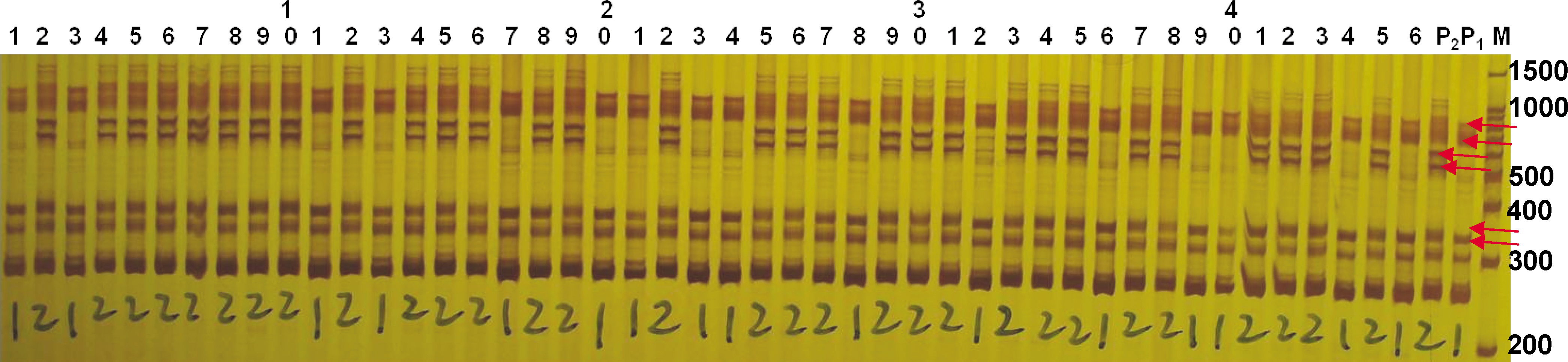
Polyacrylamide gel for the SSR marker ARS398 possessing 2 co-segregating alleles between 500 – 1000 bp in length. This image was acquired following electrophoresis with a 6% (w/v) acrylamide/bis-acrylamide (29∶1) gel at 160 V for 1 h 20 min. DNA staining, scoring techniques, and lane assignments are the same as those described in the caption for Fig. 1. This also illustrates the drawback and the limitation of this system in separation and resolution. Because of the short gel size it is difficult to separate the smaller polymorphic bands with 2–3 pb difference as indicated by lower two horizontal arrows.
The drawback is, because of its short gel size, limited in separation and resolution of PCR amplicons of specific size differences. Figure 3 shows the separation of a group of 3 co-segregating alleles of the SSR marker ARS190 ranging in approximate size of 110 – 130 bp following electrophoresis in a 9% (w/v) acrylamide/bis-acrylamide (29∶1) gel at 180 V for 1 h 40 min. The polymorphism between the groups of co-segregating alleles for ARS190 in both parental genotypes was estimated to be 2 – 3 bp. This is representative of the lower extreme of the system's resolution of small fragments possessing slight polymorphism between genotypes. Because of the size limitation and the low resolution for certain markers such as ARS190 (Figure 3), it is difficult to score correctly and could lead to errors in genotyping.
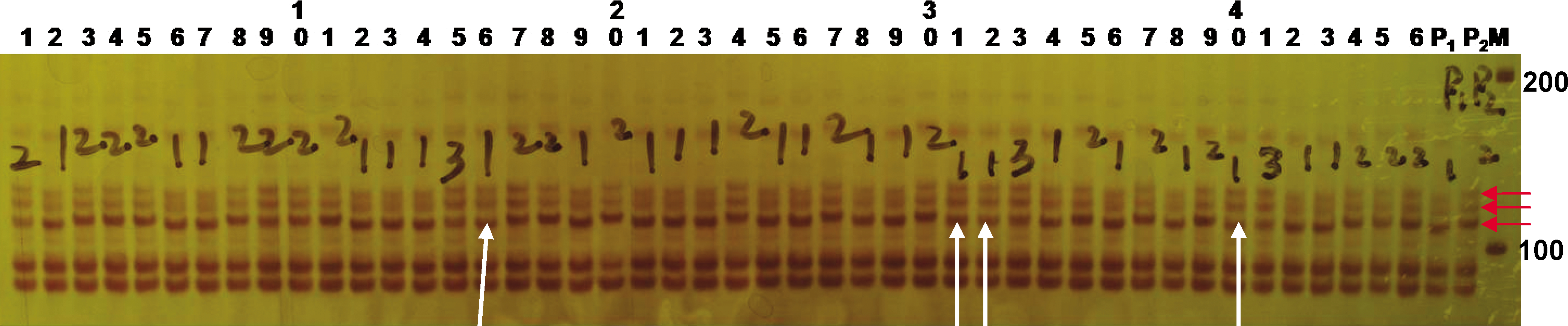
Polyacrylamide gel for the SSR marker ARS190 possessing 3 co-segregating alleles estimated at 110 - 130 bp in length. This image was acquired following electrophoresis with a 9% (w/v) acrylamide/bis-acrylamide (29:1) gel at 180 V for 1h 40 min. DNA staining, scoring techniques, and lane assignments are the same as those described in the caption for Fig. 1. The limitation of this system is difficult to separate the smaller polymorphic bands with 2-3 pb difference as indicated by vertical arrows misreading “3” to “1”.
These resolutions can be compared to other gel and staining systems. For instance, MetaPhor agarose can be used to visualize smaller DNA fragments (≤ 100 bp) as well as to resolve polymorphisms of approximately two base pairs (Reddy et al., 2001). However, this method requires the use of intercalating florescent dyes such as ethidium bromide to visualize isolated DNA fragments. This limits the overall sensitivity of similar slab gel methods in that ethidium bromide has been shown to be able to detect quantities of DNA in agarose gels as low as 1 – 3 ng/band while silver staining methods similar to those used in this protocol have been shown to be able to detect DNA in polyacrylamide gels in quantities as low as 4 – 12 pg/band (Bassam et al., 1991; Hwang et al., 2006). In addition, this silver staining method is more desirable than using intercalating fluorescent dyes such as ethidium bromide or SYBR Green, in which the mobility of DNA samples during gel electrophoresis is reduced or modified with the use of these dyes if they are incorporated into the gel prior to electrophoresis (Podzorski et al., 2006).
Given the system's use of a modified silver staining method, there is no-need of using potentially harmful intercalating fluorescent dyes, such as ethidium bromide. Wang et al. (2003) reported a similar method but in a large gel format (22 cm in length) and using ethidium bromide; thus it is a great safety improvement. The lack of such dyes, which are known to have mutagenic properties (Singer et al., 1999), prevents their possible contamination of laboratory bench surfaces and equipment thus reducing the hazards presented to users and others.
The cost of the materials needed for this system is also much less than that of other PAGE-based genotyping systems such as that described by Wang et al. (2003), which used a large gel format and required greater amount of gel materials, resulting in a cost of $2.60 per gel. Also, that system required a larger amount of PCR product (up to 30 µL) in order to achieve appropriate resolution and sensitivity than the system presented here which requires a loading volume of only 1.5 µL, therefore, decreasing PCR reaction volume from 15 to 10 µL will further reduce the cost of overall operating cost of the system. Other horizontal gel systems using standard high-melt agarose or MetaPhor agarose gels possesses similar issues in that large amounts of gel materials and PCR products are required for their use in genotyping applications given the necessity for large format gels, both resulting in increases in cost of operation.
Each apparatus used in this system is priced at less than $200 and each holds two vertical 52-sample gels. The cost of materials for the nondenaturing polyacrylamide gel used in the system is approximately $0.53 per gel, with each gel producing a maximum of 52 data points. Also, the cost of the materials for the silver staining method presented here is estimated at $0.37 per gel. This results in a total cost of $0.018 per data point, excluding PCR and DNA preparation costs. In addition, given the lack of necessity for ultraviolet light and filters to visualize the DNA fragments in the gels, a molecular imager is not required and a standard digital camera may be used. This reduces the required equipment costs and makes this method more cost-effective and easy to operate.
The throughput of this system in genotyping applications in peanut has been found to be sufficient. Presently, our laboratory operates 8 of these electrophoresis systems. With this, 1 – 2 people can consistently run a total of 24 to 32 gels per day (2 gels per apparatus, 12 to 16 gels per run) with a complete run time of approximately 4 hours, including electrophoresis and staining times (PCR preparation and running times are not included here but could be well planned and done during the electrophoresis). Based on this rate, 1 – 2 people can generate more than 1200 – 1600 data points in 8 hours, depending on the number of samples loaded into each gel. This throughput is made possible largely in part by the modified silver staining method based on that of Zhang et al. (2000), and the rate and low cost at which staining and electrophoresis can be performed. This staining protocol can be completed in approximately 30 min. This is much faster than the time required for other methods, which may require more than 2 hours to complete, with equivalent levels of sensitivity (Beidler et al., 1982; Bassam et al., 1991; Zhang et al., 2000; Creste et al., 2001; Wang et al., 2003).
The drawback of this system includes the small gel-size and short run time for some markers resulting in insufficient separation and difficulty to score the genotypes correctly. Silver stain in some cases will produce strong background and interfere with the genotype scores. With some practice and adjustment in gel concentration, voltage, run time and staining, these issues could be improved. Given the advantages of simplicity, sensitivity, and the lack of necessity for expensive laboratory equipment, such as molecular imagers, combined with the low cost and rapid rate of data point collection, this system could be a good alternative along with other genotyping methods.
Acknowledgements
We thank Billy Wilson for technical assistance in the field and the laboratory. This research was partially supported by the Peanut Foundation, and the Georgia Peanut Commodity Commission. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Literature Cited
Bassam B. J Caetano-Anollés G and Gresshoff P. M 1991 Fast and sensitive silver staining of DNA in polyacrylamide gels Ana. Biochem. 196 : 80 – 83 .
Beidler J. L Hilliard P. R and Rill R. L 1982 Ultrasensitive staining of nucleic acids with silver Ana. Biochem. 126 : 374 – 380 .
Creste S Neto A. T and Figueira A 2001 Detection of single sequence repeat polymorphisms in denaturing polyacrylamide sequencing gels by silver staining Plant Mol. Biol. Reporter. 19 : 299 – 306 .
Guo B. Z Chen C. Y Chu Y Holbrook C. C Ozias-Akins P and Stalker H. T 2011 Advances in genetics and genomics for sustainable peanut production In Benkeblia N (ed.) , Sustainable Agriculture and New Biotechnologies, CRC Press , Boca Raton, FL , (in press, July 2011) .
He G Woullard F. E Marong I and Guo B. Z 2006 Transferability of soybean SSR markers in peanut (Arachis hypogaea L.) Peanut Sci. 33 : 22 – 28 .
Holbrook C. C Timper P Dong W Kvien C. K and Culbreath A. K 2008 Development of near-isogenic peanut lines with and without resistance to the peanut root-knot nematode Crop Sci. 48 : 194 – 198 .
Hong Y Chen X Liang X Liu H Zhou G Li S Wen S Holbrook C and Guo B. Z 2010 A SSR-based composite genetic linkage map for the cultivated peanut (Arachis hypogaea L.) genome BMC Plant Biol. 10 : 17 .
Hwang S. Y Jin L. T Yoo G. S and Choi J. K 2006 Silver staining method for DNA in polyacrylamide gels using eriochrome black T as a silver-ion sensitizer Electrophoresis. 27 : 1744 – 1748 .
Isleib T. G Holbrook C. C and Gorbet D. W 2001 Use of Arachis spp. plant introductions in peanut cultivar development Peanut Sci. 28 : 96 – 113 .
Isleib T. G Young C. T and Knauft D. A 1996 Fatty acid genotypes of five Virginia type cultivars Crop Sci. 36 : 556 – 558 .
Knauft D. A Moore K and Gorbet D. W 1993 Further studies on the inheritance of fatty acid composition in peanut Peanut Sci. 20 : 74 – 76 .
Knauft D. A and Wynne J. C 1995 Peanut breeding and genetics Adv. Agron. 55 : 393 – 445 .
Liang X Chen X Hong Y Liu H Zhou G Li S and Guo B. Z 2009 Utility of EST-derived SSR in cultivated peanut (Arachis hypogaea L.) and Arachis wild species BMC Plant Biol. 9 : 35 .
Podzorski R. P Loeffelholz M and Hayden R. T 2006 . Detection and characterization of molecular amplification products: agarose gel electrophoresis, southern blot hybridization, restriction enzyme digest analysis, and enzyme-linked immunoassay, pp. 243 – 263 In Tang Y and Stratton C. W (eds.), Advanced Techniques in Diagnostic Microbiology Springer , New York .
Qin H Chen C Feng S Guo Y Knapp S Culbreath A He G Wang M. L Zhang X Holbrook C. C Ozias-Akins P Liang X and Guo B. Z An integrated genetic linkage map of cultivated peanut (Arachis hypogaea L.) constructed from two RIL populations Theoretic and Applied Genetics (submitted)
Rahman M. H Jaquish B and Khasa P. D 2000 Optimization of PCR protocol in microsatellite analysis with silver and SYBR® stains Plant Mol. Biol. Reporter. 18 : 339 – 348 .
Reddy O. U. K Pepper A. E Abdurakhmonov I Saha S Jenkins J. N Brooks T Bolek Y and El-Zik K. M 2001 New dinucleotide and trinucleotide microsatellite marker resources for cotton genome research J. Cotton Sci. 5 : 103 – 113 .
Simpson C. E and Starr J. L 2001 Registration of ‘COAN’ peanut Crop Sci. 41 : 918 .
Singer V. L Lawlor T. E and Yue S 1999 Comparison of SYBR® Green I nucleic acid gel stain mutagenicity and ethidium bromide mutagenicity in the Salmonella/mammalian microsome reverse mutation assay (Ames test) Mutation Res. 439 : 37 – 47 .
Varshney R. K Bertioli D. J Moretzsohn M. C Vadez V Krishnamurthy L Aruna R Nigam S. N Moss B. J Seetha K Ravi K He G Knapp S. J and Hoisington D. A 2009 The first SSR-based genetic linkage map for cultivated groundnut (Arachis hypogaea L.) Theor. Appl. Genet. 118 : 729 – 39 .
Wang D Shi J Carlson S. R Cregan P. B Ward R. W and Diers B. W 2003 A low-cost, high-throughput polyacrylamide gel electrophoresis system for genotyping with microsatellite DNA markers Crop Sci. 43 : 1828 – 1832 .
Wang M. L Barkley N. A and Jenkins T. M 2009 Microsatellite markers in plants and insects. Part I: Applications of biotechnology Genes, Genomes and Genomics 3 : 54 – 67 .
Zhang J Wu Y. T Guo W. Z and Zhang T. Z 2000 Fast screening of SSR markers in cotton with PAGE/silver staining Cotton Sci. Sinica. 12 : 267 – 269 .
Notes
- USDA-ARS, Crop Protection and Management Research Unit, Tifton, GA 31793
- Louisiana State University, Department of Plant Pathology and Crop Physiology, Baton Rouge, LA 70803
- University of Georgia, Department of Plant Pathology, Tifton, GA 31793
- Hubei Academy of Agricultural Sciences, Oil Crop Research Institute, Wuhan 430064, China
- USDA-ARS, National Peanut Research Laboratory, Dawson, GA 39842
- USDA-ARS, Plant Genetic Resources Conservation Unit, Griffin, GA 30223 *Corresponding author e-mail: baozhu.guo@ars.usda.gov
Author Affiliations