

Introduction
The identification of true hybrids is important for peanut breeding programs. Crossing involves removal of ten stamens in the evening, followed by cross-pollination in the morning. It is easy to miss a stamen, which can remain hidden in the keel and cause self pollination. In addition, the peanut inflorescence is compound, with two or three flowers produced at the same axil within approximately one week. Selfed flowers must be removed early in the morning, and if any are missed or pulled late, a selfed peg may emerge at the same site as a hybrid peg or where an unsuccessful attempt at cross-pollination had occurred.
Several methods are used to distinguish hybrid versus selfed progenies, including observing morphological differences among progenies, segregation for disease resistance or differences in the oleic:linoleic ratio (López and Burow, unpublished results). Identifying hybrids in the F1 generation can be difficult because the F1 may not be readily distinguishable from the parents, especially in the greenhouse where plants cannot grow to full size due to limited space. In the field, it is often possible to distinguish F2 plants by segregation for morphological traits. However, this may not be the case for closely-related parents and may not be useful in the case of attempted three-way crosses, where failure to cross hybrids produces segregating progeny. In addition, planting of F2 plants to identify hybrids by appearance is an inefficient use of field space and labor. Finally, discovery of selfs typically occurs a year after the crosses are made, resulting in potential delays to improvement programs.
Identification of hybrids can be performed through use of DNA markers. Codominant markers are preferable because they produce different alleles (markers) for each parent, and F1 hybrids will possess an allele from each parent. Of the major DNA marker types, restriction fragment length polymorphism (RFLP) and simple sequence repeat (SSR) markers are usually codominant. The SSR-based markers require smaller quantities of DNA than do RFLP-based markers (Powell et al., 1996), and analysis by SSR markers is quicker and does not involve the use of radioisotopes. Random amplified polymorphic DNA (RAPD) markers are often not reproducible, and are dominant in most cases and therefore markers from the female parent cannot detect heterozygotes in the F1 generation (Burow and Blake, 1998), although segregation of marker patterns in the F2 generation can identify hybrids. Use of SSR markers has allowed distinguishing among accessions of the cultivated peanut species, A. hypogaea L. (Kottapalli et al., 2007); this was not possible with RFLP or RAPD markers (Kochert et al., 1991; Halward et al., 1991). The use of microsatellite markers for assessing true hybrids is common in tomato (Smith and Register, 1998), maize (Salgado et al., 2006), rice (Yashitola et al., 2002), and cotton (Dongre and Parkhi, 2005).
Currently, several separation methods are employed to determine the length of amplification products; among the methods are agarose gels and non-denaturing polyacrylamide gel electrophoresis (PAGE) (Ogden and Adams, 1987). Agarose gels are simple to use, but their limited resolution means that small differences in repeat length are not observable. Specialized agarose gels offer improved resolution but are expensive (Wang et al., 2003). PAGE gels have relatively high resolution but require expensive vertical gel units and are tedious to pour.
This paper describes an inexpensive and simple method (H-PAGE) for identification of peanut hybrids in the F1 or F2 generation. SSR markers were used to distinguish parents of cultivated × cultivated crosses, or of crosses involving one cultivated and one wild species introgression line parent. After this, putative progeny were tested for presence of the male parent allele. Additionally, the use of horizontal polyacrylamide gel electrophoresis provided a simple and inexpensive method of separation of alleles differing by a few base pairs.
Materials and Methods
Plant Materials
Experimental materials were comprised of F1 and F2 populations for development of heat stress-tolerant and leaf spot-resistant lines, respectively. Lines developed for heat stress tolerance included putative F1 plants derived from A. hypogaea L. parents ICGS-76 (Nigam et al., 1991) × Tamrun OL02 (Simpson et al., 2006), ICGV-87157 (Nigam et al., 1992) × Tamrun OL02, and ICGS-76 × Spanco (Kirby et al., 1989). Five populations were developed for leaf spot resistance; each population had one interspecifically - derived breeding line as a parent. The breeding line parents were BC3F6 progeny of the cross (Florunner × TxAG-6), where TxAG-6 was a synthetic amphidiploid (Simpson, 1991). The five crosses were: 41-10-01-03 × Tamrun OL02, Tamrun OL02 × 43-09-03-02, 63-04-02-02 × Tamrun OL02, 55-437 (Bockelee, 1983) × 43-09-03-02, and 55-437 × 45-04-02-01. Crossing was carried out in 2005 and 2006 at the Texas Tech University greenhouse by hand emasculation in the evening, followed by artificial pollination in the morning. Populations derived for leafspot resistance were advanced to the F2 generation by self pollination.
Sample Collection and Dna Isolation
All parents and F1 and F2 progenies were grown in potting soil (Sunshine SB-300) in plastic trays in the greenhouse at the Texas Agricultural Experimental Station (Lubbock, TX) greenhouse for 25 to 28 days to allow for collection of tissue. After confirmation of hybridization, plants were transplanted to larger pots for seed production. For putative F1 crosses, tissue was collected from only one plant per pod. For putative F2 populations, from 12 to 36 seeds were sown from each F1, and six randomly selected plants from each cross were used for marker analysis.
Unopened tetrafoliate leaves from 20 to 25 day-old parents or putative hybrids were used for DNA isolation. Leaves were stored at -80°C, or were collected fresh. Leaves were ground in a mortar and pestle using liquid nitrogen. Genomic DNA was isolated as per Dellaporta et al. (1983) or using the Qiagen DNeasy kit (Qiagen Inc., Valencia, CA). DNA concentration was determined by agarose gel electrophoresis and comparison of staining intensity with phage DNA standards (Promega Inc., Madison, WI) loaded at 300, 200, 100, and 30 ng per lane. Peanut DNA samples were diluted using TE buffer to a final concentration of 30 ng μl-1.
PCR Amplification and Detection of Polymorphisms
A total of 24 oligonucleotide primer pairs flanking microsatellite repeat sequences were used in the present study. Fifteen primer pairs (PM3, PM15, PM32, PM35, PM36, PM45, PM50, PM53, PM65, PM137, PM145, PM183, PM188, PM200, and PM210) were from the published sequences of He et al. (2005); six primer pairs (Ah41, Ah75, Ah193, Ah229, Ah522, and Ah558) were from Moretzsohn et al. (2004), and PGS14F05, PGS04D04, PGS12A07, and PGS14E10 were from Ferguson et al. (2004).
The PCR reaction condition used was as follows: DNA samples (30 ng) were amplified in a 10 μl reaction volume containing 1× Polymerase Chain Reaction buffer (10 mM Tris HCl pH 8.3, 50 mM KCl, 0.1% Triton X-100, and 0.01% gelatin), 0.2 mM each of the four dNTPs, 2 mM MgCl2, 0.5 μM of each forward and reverse primer (synthesized by Integrated DNA Technologies, Coralville, IA), and 0.5 U of Hot Start Taq polymerase (Qiagen Inc, Valencia CA.) PCR was performed in a PTC-200 thermal cycler (Biorad Inc., Hercules CA) with an initial denaturation at 94°C for 3 min, then 19 cycles using a touch-down strategy (Mellersh and Sampson, 1993) (initial cycle 94°C for 30 s, 63°C for 30 s, 72°C for 1 min, lowering the annealing temperature for each cycle by 0.5°C during the following 19 cycles), followed by 19 cycles of 94°C for 15 s, 55°C for 30 s and 72°C for 1 min. Cycling was followed by a final extension at 72°C for 10 min, and a soak at 4°C.
Electrophoresis
Non-denaturing polyacrylamide gels were cast in a horizontal gel casting plate designed for agarose gels. A 6% polyacrylamide gel was prepared using an acrylamide/bisacrylamide ratio of 19:1, 0.5× TBE (Tris boric acid ethylenediamine tetraacetic acid) buffer (Sambrook et al., 1989), 0.1% ammonium persulfate (APS), and 8.33% tetramethyl ethylenediamine (TEMED). Immediately after addition of APS, 70 ml of the gel solution was poured to a depth of approximately 2.5 mm directly into the gel casting plate (16 × 14 cm) blocked at the end with baffles and combs inserted into the solution. The plate with gel solution was then kept at room temperature for approximately 2 to 3 minutes to allow polymerization. After polymerization, the gel plate was stored under pre-chilled buffer (0.5× TBE), and after removing combs and baffles, samples were loaded as for agarose gels. Care was taken to avoid overexposure of the gel to air to prevent shrinkage. Five μl of Type IV gel loading buffer described by Sambrook et al. (1989) were added to the 10 μl PCR product, and 4 μl of the sample were loaded on the gel. Gels were run in a submarine horizontal electrophoresis unit (CBS Scientific, Del Mar CA ) for 2.5 hrs at 3 V cm-1. The covered gel unit was covered with ice packs for better resolution of the amplicons.
After electrophoresis, the gel was stained in 500 ml of water containing 15 μl ethidium bromide (100 mg/ml) for 15 to 20 minutes, followed by destaining for 15 minutes in distilled water. The staining solution was stored in the dark and could be used up to three times. Alternatively, ethidium bromide (25 μl for one liter of running buffer) could be added to the running buffer. The same running buffer was reused two additional times effectively. After staining, the gel was visualized either on a UV transilluminator (Model FBTV-816, Fisher Biotech, Pittsburgh PA), photographed using a Kodak DC-290 camera with a deep yellow 15 filter (Tiffen, Inc., Glendale, CA) connected to a PC running Slackware Linux v 10.2 (), and images visualized using the included digikam software, or using an Alpha Imager TM 2200 (AlphaInnotech Inc., San Leandro, CA) gel documentation system. The H-PAGE gels were also compared with 4% standard agarose (Fisher Biotech) and 4% SFR (Super Fine Resolution) agarose (Amresco, Solon, OH) gels using 25 or 100 bp ladder DNA molecular weight markers (Promega Corp, Madison, WI) to test resolution.
Results and Discussion
A method for identification of true hybrids in peanut was developed. To our knowledge, this is the first use of DNA markers for this purpose in cultivated peanut. In this paper, we detail a new, simple, low cost method which could be used in peanut breeding programs worldwide.
DNA Isolation and Quality
Chemotypic heterogeneity among species may not allow optimal DNA yield with a single isolation protocol. Thus, even closely-related species may require different DNA extraction protocols (Loomis, 1974; Weishing et al., 1995). Two DNA isolation methods were examined. The Dellaporta method (Dellaporta et al., 1983) was found to be satisfactory for crosses involving cultivated genotypes but not for wild species. The concentration of DNA obtained from cultivated crosses by the Dellaporta method ranged between 600 and 1030 ng μl-1. DNA quality was poor from crosses involving wild species introgression lines using the Dellaporta method. The DNA obtained was viscous and only 40% of the samples were amplified by PCR. To avoid this problem, the Qiagen DNEasy kit was used to isolate DNA from the crosses involving wild species. Compared to the Qiagen kit, the Dellaporta method is inexpensive and the materials cost per sample is approximately $0.20.
SSR Polymorphism and Identification of True Hybrids
Of the 24 microsatellite loci analyzed, eight were observed to be polymorphic (PM3, PM32, PM50, PM137, PM188, PM210, Ah193 and PGS12A07) for the lines screened. Four SSR markers (PM210, PM42, PM3 and PGS12A07) showed clear polymorphism for most of the crosses (Table 1). Marker patterns observed in progeny were consistent with what would be expected based on parental allele sizes.
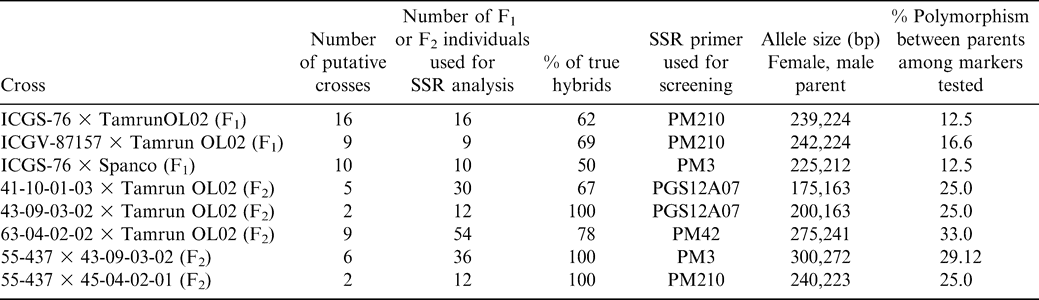
Crosses tested for hybrid production and results of SSR analysis.
The present study revealed 14 and 27% polymorphism in cultivated × cultivated and interspecific crosses, respectively. Although polymorphism was lower in cultivated × cultivated crosses than using interspecifically-derived lines as one parent, the set of 24 primer pairs used was adequate for identification of polymorphism in all crosses used (see Table 1). In the putative F2 populations, DNA was analyzed from six F2 plants derived from each F1 (Figure 1) to give a less than 2% chance of falsely classifying the cross as a self if it was actually a hybrid. This was a conservative estimate, using only heterozygotes as proof of hybridization. Male parent patterns could also be considered to be evidence of hybrids, in which case only three individuals would need to be tested for 98% confidence of correctly identifying hybrids. However, the not-uncommon error of reversing male and female parents when writing crossing tags would allow for a higher, but unspecified, error rate. The presence of the male parent allele, either in the form of the heterozygote or male homozygote in any one of six samples indicated that the original cross succeeded, and all progeny of that F1 were hybrids. Testing of F1 plants is much more efficient than testing F2 plants, but the experiment demonstrated that if, for some reason, tissue is not available from the F1 plants, the F2 generation can be tested. Overall, it was found that 70% of the putative hybrids were true hybrids (Table 1).
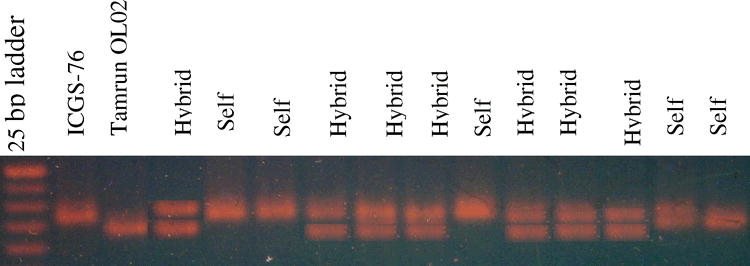
Microsatellite marker survey for detecting true F1 hybrids. Polymorphism between the cross ICGS-76 × Tamrun OL02, and putative F1 hybrids, using primer pair PM 210. Marker sizes are 300, 275, 250, 225, and 200 bp.
Horizontal Polyacrylamide Gel Electrophoresis (h-Page)
Horizontal PAGE has good resolving potential for distinguishing the heterozygote from the homozygote (Figures 1 and 2). Agarose gels are easy to prepare, but their limited resolution means that small differences in repeat length are not observable (Figure 3). Specialized super fine resolution (SFR) agarose gels have been used to separate alleles of microsatellite markers, but the cost is five times more than that of nondenaturing polyacrylamide gels (Wang et al., 2003). PAGE gels have relatively high resolution, but require expensive vertical gel units and are tedious to pour. The main advantages of the H-PAGE method is that gel preparation is as easy and rapid as agarose gel preparation. The horizontal polyacrylamide gels can be run on electrophoretic units designed for agarose gels, eliminating the time-consuming gel casting procedure for vertical gels. Use of ethidium bromide is simpler and cheaper than silver staining procedures used for PAGE. The resolved bands were visualized clearly after ethidium bromide staining. The ethidium bromide staining requires 30 minutes but this step can be eliminated if the ethidium bromide is mixed with running buffer in the electrophoresis tank. Finally, horizontal gel units are significantly cheaper than vertical units.
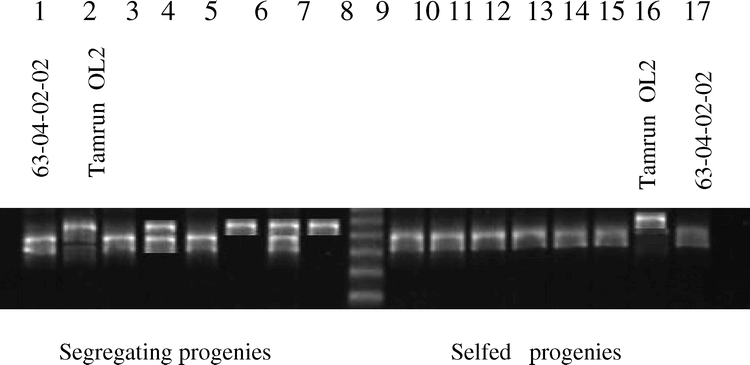
F2 (63-04-02-02 × Tamrun OL02) progeny survey using microsatellite primer pair PM42. Polymorphism was evident among F2 plants derived from one F1 (lanes 3-8). Lanes 10-15 are F2 progeny derived from a different F1, demonstrating selfed progenies. Molecular weight marker (lane 9) is a 25 base pair DNA ladder; marker sizes are 275, 250, 225, 200, and 175 bp.

Separation of DNA by various electrophoretic methods. A. Separation of different DNA molecular weight markers, a 25 bp ladder (lane 1) and 100 bp ladder (lane 2) were fractionated electrophoretically on 4% agarose, 4% SFR agarose or 6% horizontal polyacrylamide gel electrophoresis (H-PAGE) up to 1.5 hrs under the same electrophoretic conditions. Gels were stained with ethidium bromide as described in materials and methods. B. Comparison of SSR allele (PM210) separation of peanut F1 hybrids and their parents (ICGS-76 × Tamrun OL02) using standard agarose and the H-PAGE system.
Use of the Kodak DC-290 camera also allows for inexpensive visualization of results. This camera, or similar models, is inexpensive, and has the ability to take close-up photos. Connection to a personal computer running Slackware Linux provided all the needed software at no cost. With the help of the free Image J software (Rasband, 1997), it was possible to set up a photographic station capable of photographing, storing, printing gels and determining the molecular weights of bands (Figure 4).
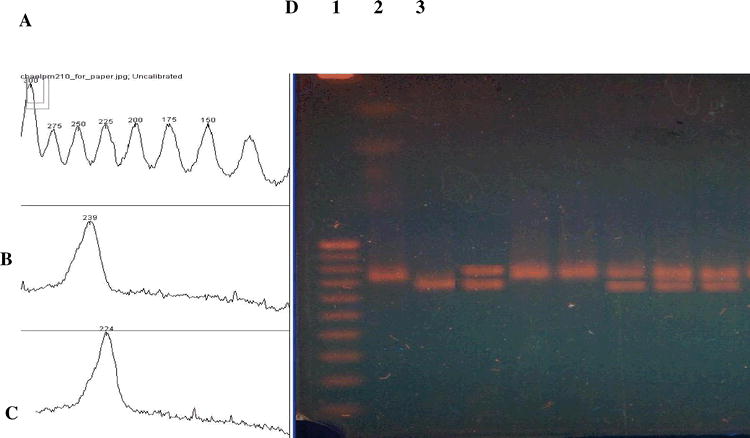
Determination of the size of SSR allele PM210 using Image J software. (A) Size (bp) of standards (Panel D, lane 1), (B). Size determination of one allele (panel D, lane 2), (C). Size of the other allele (panel D lane 3), (D) Photograph of the gel. Size markers in lane 1 are a 25 bp ladder (sizes 300 bp to 50 bp are shown); lanes 2 and 3 are amplified products from ICGS-76 and Tamrun OL02, respectively.
This system is ideal for small-scale breeding or newly-established laboratories with very limited facilities. For the Dellaporta DNA extraction method, a low-speed (3500 rpm) centrifuge, microcentrifuge, and heated conventional water bath are needed, but for the Qiagen DNA Easy kit protocol, a micro centrifuge and heated water bath are the only major pieces of equipment needed for DNA extraction. For detection, a UV transilluminator to visualize the DNA fragments is needed, and a camera is desirable to reproduce images. An inexpensive PC and printer using open source software can be used for long-term storage of images and printing of results. Also, the same horizontal gel unit can be used for agarose gel electrophoresis. The cost of using this method is low as this method does not require any sophisticated vertical apparatus. The gel ingredients cost less than a dollar, and a gel can be used to obtain 52 data points (two 26 well combs in 16 × 14 cm gel plate) without multiplexing. This system may be compared favorably with high-resolution agarose gels that are widely used in many laboratories for genotyping with microsatellite markers. This method is cheaper than the high-resolution SFR or Metaphor (Lonza Inc, Rockland, ME) agaroses used for SSR work, and amplified bands are clearer and sharper than those on SFR agarose gels. Currently we are using this method to enrich the tetraploid peanut map using microsatellite markers.
Conclusion
The horizontal PAGE method was used successfully to verify hybrids in F1 and F2 populations of peanuts using microsatellite markers. The high discriminating power of SSR markers and inexpensive setup should allow this to be affordable for many peanut breeding laboratories.
Acknowledgments
The authors would like to express thanks to Jamie Ayers and Yolanda López for assistance with greenhouse work. Funding for these studies was provided by the National Peanut Board, the Southwest Consortium for Plant Genetics and Water Resources (#2004-34186-14533), and the Peanut Collaborative Research Support Project.
Literature Cited
Bockelee M. A. 1983 The different varieties of groundnut. Geographical and climatic distribution, availability. Technical sheet for groundnut variety 55-437. Oléagineaux 38: 80.
Burow M. D. and Blake T. K. 1998 Molecular tools for the study of complex traits. 13- 29 In Paterson A. H. Molecular analysis of complex traits CRC Press Boca Raton.
Dellaporta S. L., Wood J., and Hicks J. B. 1983 A plant DNA minipreparation: Version II. Plant Mol. Biol. Rep 1: 19- 21.
Dongre A. and Pakri V. 2005 Identification of cotton hybrids through the combination of PCR based RAPD, ISSR and microsatellite markers. J. Plant Biochem. Biotech 14: 53- 55.
Ferguson M. E., Burow M. D., Schulze S. R., Bramel P. J., Paterson A. H., Kresovich S., and Mitchell S. 2004 Microsatellite identification and characterization in peanut (A. hypogaea L.). Theor. Appl. Genet 108: 1064- 1070.
Halward T., Stalker H. T., Larue E. A., and Kochert G. 1991 Use of single-primer DNA amplifications in genetic studies of peanut (Arachis hypogea L.). Plant Mol. Biol 18: 318- 325.
He G. H., Meng R., Gao H., Guo B., Gao G., Newman M., Pittman R. N., and Prakash C. S. 2005 Simple sequence repeat markers for botanical varieties of cultivated peanut (Arachis hypogaea L.). Euphytica 142: 131- 136.
Kochert G., Halward T., Branch W. D., and Simpson C. E. 1991 RFLP variability in peanut (Arachis hypogea L.) cultivars and wild species. Theor. Appl. Genet 81: 565- 570.
Kottapalli K. R., Burow M. D., Burow G., Burke J., and Puppala N. 2007 Molecular characterization of the U.S. peanut mini core collection using microsatellite markers. Crop Sci 47: 1718- 1727.
Loomis M. D. 1974 Overcoming problems of phenolics in the isolation of plant enzymes and organelles. Meth. Enzymol 31: 528- 545.
Mellersh C. and Sampson J. 1993 Simplifying detection of microsatellite length polymorphisms. BioTechniques 15: 582- 584.
Moretzsohn M. C., Hopkins M. S., Mitchell S. E., Kresovich S., Valls J. F. M., and Ferreira M. E. 2004 Genetic diversity of peanut (Arachis hypogaea) and its wild relatives based on the analysis of hypervariable regions of the genome. BMC Plant Biol 4: 11.
Nigam S. N., Dwivedi S. L., Rao Y. L., and Gibbons R. W. 1991 Registration of 'ICGV 87141' peanut. Crop Sci 31: 1096.
Nigam S. N., Reddy L. J., Subrahmanyam P., Reddy A. G. S., McDonald D., and Gibbons R. W. 1992 Registration of ICGV 87157, an elite peanut germplasm with multiple resistance to diseases. Crop Sci 32: 837.
Ogden R. C. and Adams D. A. 1987 Electrophoresis in agarose and acrylamide gels. Meth Enz 152: 61- 87.
Powell W., Machray G., and Provan J. 1996 Polymorphism revealed by simple sequence repeats. Trends Plant Sci 1: 215- 222.
Salgado K. C. P. C., Vieira M. G. G. C., Pinho éV. R. V., Guimarães C. T., Pinho R. G. V., and Souza L. V. 2006 Genetic purity certificate in seeds of hybrid maize using molecular markers. Revista Brasileira de Sementes 28: 169- 175.
Simpson C. E. 1991 Pathways for introgression of pest resistance in to Arachis hypogea L. Peanut Sci 18: 22- 26.
Simpson C. E., Baring M. R., Schubert A. M., Black M. C., Melour H. A., and Lopez Y. 2006 Registration of 'Tamrun OL 02' peanut. Crop Sci 46: 1813- 1814.
Smith J. S. C. and Register J. C. 1998 Genetic purity and testing technologies for seed quality: a company perspective. Seed Sci. Res 8: 285- 293.
Wang D., Shi J., Carlson S. R., Cregan P. B., Ward R. W., and Diers B. W. 2003 A low-cost, high-throughput polyacrylamide gel electrophoresis system for genotyping with microsatellite DNA markers. Crop Sci 43: 1828- 1832.
Yashitola J., Thirumurugan T., Sundaram R. M., Naserullah M. K., Ramesha M. S., Sarma N. P., and Sonti R. V. 2002 Assessment of purity of rice hybrids using microsatellite and STS markers. Crop Sci 42: 1369- 1373.
Notes
- First and second authors: Res. Agron. , USDA-ARS and Agric. Res. Statistician; Coastal Plain Exp. Sta., Tifton, GA 31793-0748. Third author: President and CEO, Hebert Green AgroEcology; Asheville, NC 28801. [^]
- Texas Tech University, Department of Plant and Soil Science, Lubbock, TX 79409 [^]
- Savanna Agricultural Research Institute, Tamale, Ghana [^]
- Central Research Institute for Jute and Allied Fibers, Kolkata-700 120, India [^]
- Texas A&M University Agricultural Experiment Station, Stephenville, TX 76401 [^]
- USDA-ARS, Cropping Systems Research Lab, Lubbock, Texas 79415 [^]
- New Mexico State University, Agricultural Science Center, Clovis, NM 88101 [^]
- Texas A&M Agricultural Experiment Station, 1102 East FM 1294, Lubbock, TX 79403 [^] *Corresponding author, address: Texas Agricultural Experiment Station, 1102 East FM 1294, Lubbock, TX 79403, Phone: (806)-746-6101, FAX: (806)-746-6528, E-mail: mburow@tamu.edu.
Author Affiliations