

Introduction
Selection of desirable agronomic traits in plant breeding programs has traditionally been performed at the phenotypic level which requires first growing plants either in a field or greenhouse and then assessing them for traits such as disease resistance, drought tolerance, or yield parameters. The process is time consuming and often limited by growing seasons or available greenhouse space. Traditional testing of plants in the greenhouse or field for disease resistance may result in the loss of plant material and prevention of further use for breeding programs, although for some crops such as peanut, non-destructive tests for fungal disease resistance have been developed for use in greenhouses (Pataky et al., 1983; Melouk et al., 1992).
Molecular techniques have now been developed to assess plant species at the genotypic level through Marker Assisted Selection (MAS). MAS can greatly improve the efficiency of cultivar development. One advantage molecular markers have over the traditionally used phenotypic markers is that they are unaffected by the environmental conditions in which plants are grown and are not dependent upon plant growth stage which eliminates the restriction of growing seasons. In general, MAS is a non-destructive process that allows for the identification of plants with desirable traits for advancement in a breeding program. Techniques which are common in developing molecular markers include random-amplified polymorphic DNAs (RAPDs), restriction fragment length polymorphisms (RFLPs), sequence characterized amplified regions (SCARs), amplified polymorphic DNAs (AFLPs), and simple sequence repeats (SSRs) (Mohan et al., 1997; Charcosset and Moreau, 2004; Collard et al., 2005).
Marker assisted selection in plant breeding programs is not without limitations, however. The availability of markers for a specific crop can limit MAS success in breeding programs. Important agronomic traits such as yield and yield components, plant height, maturity, and disease resistance are often controlled by several genes or quantitative trait loci (QTL). However, the number of markers associated with QTLs is rapidly increasing for crop plants. For peanut alone, molecular markers have been identified for maturity (Bland and Lax, 2000), nematode resistance (Burrow et al., 1996; Garcia et al., 1996), late leaf spot resistance (Luo et al., 2005), and resistance to the aphid vector of groundnut rosette disease, Aphis craccivora (Herselman et al., 2004).
The ability to quickly and economically extract quality DNA from large populations of plants can also limit the usefulness of MAS to plant breeders. Methods are available for isolation of plant genomic DNA, but many are species-specific and involve taking tissue from whole plants. MAS is most efficient when DNA can be taken from seed in a non-destructive manner so that positively identified samples can then be used to produce mature plants for breeding programs. To this end, methods for extracting DNA directly from seed have been developed for wheat (Hill-Ambroz et al., 2002), barley (von Post et al., 2003), soybean (Bolton et al., 2005), ryegrass (Sweeney and Danneberger, 1997), and turfgrass (Sweeney et al., 1996). For peanuts, however, extraction of quality DNA from peanut is problematic because peanuts contain a high percentage of phenolic compounds and polysaccharides which inhibit many molecular techniques (Demeke and Adams, 1992; Pandey et al., 1996). Methods have been developed to purify DNA from peanut leaves (Paik-Ro et al., 1992; Choi et al., 1999; Sharma et al., 2000) and one report of using commercially available extraction kits to extract DNA from whole peanut seed has been published (Hird et al., 2003). However, these methods do not allow for low cost and non-destructive extraction of DNA from peanut seed which is necessary to realize the full potential of MAS in peanut breeding programs. Thus, the objective of this research was to develop a non-destructive procedure to extract high quality DNA directly from peanut seed suitable for use in PCR based applications.
Materials and Methods
Plant Material . Dry seed from all four cultivated peanut market types were examined. Runner market types included cultivars Okrun, Florunner, Flavor Runner 458, Southwest Runner, Georgia Green, Tamrun 98, Georgia High Oleic, COAN, Hull, Norden, DP-2 and breeding lines UF98326, 920L19-8-1-1, TX901338-2, TX92622-66, TX931019-39, TX961507, TX961678, TX961709, TX971738, TX971783, TX994313, TX994374 and TX997720. Valencia market types included cultivar Valencia C and breeding lines Grif 13826, Grif 14057, PI 501983, PI 501996, PI 502009 and PI 502154. The Spanish cultivar included was Tamspan 90. Virginia market types included cultivars Early Bunch, Jupiter, NC7, NC12C, Perry and breeding lines GPNCWS-12, GPNCWS-15, N96076L, NO3023EF, NO3076FT, NO3079FT, NO3081T, NO30804FT, NO3085FT, NO3086FT, NO3088T, NO3089T, NO3090T and NO4052FCSmT. Additionally, dry seed from transgenic peanut lines 487, 540, and 654 containing antifungal transgenes (Chenault et al., 2002) were tested.
Extraction Method . To extract genomic DNA from peanut seed (summarized in Table 1), a small section was removed (approximately 0.02 g) from the distal end of the seed being careful not to disturb the embryo if subsequent germination was desired. The tissue was placed in a 1.5 mL microfuge tube and manually ground with a small plastic pestle in 400 µL extraction buffer (Table 1) until no solid seed portion remains and a milky-white solution or paste was formed. One-half vol of 20% SDS was added, vortexed well, placed at 65°C for 10 min and mixed by inversion twice during incubation period. After incubation, 1/3 vol of 5 M KAc was added and followed by an incubation on ice for 20 min. Samples were centrifuged at 15000 × g for 20 min at room temperature to pellet undesired cellular debris and the supernatant was removed and placed in a clean 1.5 mL centrifuge tube without disturbing or collecting any interface material. If supernatant was colorless and contained no particulate matter (otherwise, incubation with SDS as before was repeated), DNA was precipitated by adding 1 vol of room temperature isopropanol and mixed by inversion or vortexing. Samples were incubated at −20°C for at least 30 min (may have been left overnight at −20°C if desired). After incubation, samples were centrifuged at 15000 × g for 20 min to pellet DNA. Supernatant was removed by pipeting or pouring off and pellet was dissolved in 400 µl of Solution A (Table 1) and vortexed well. [Note: At this point in the procedure, an incubation at 65°C may be required to completely dissolve pellet] After pellet was dissolved, samples were centrifuged at 15000 × g for 20 min at room temperature to pellet any remaining particulate matter and supernatant was collected and placed into a clean 1.5 mL centrifuge tube. Organic material was then removed from the DNA mixture by extracting with 1 vol phenol∶chloroform∶isoamyl alcohol [25∶24∶1] and vortexing well until layers mixed completely. Samples were centrifuged at 15000 × g for 5 min at room temperature to separate layers and the aqueous phase (top layer) was collected, being careful not to disturb or collect interface material, and placed supernatant into a clean 1.5 mL centrifuge tube. Samples were then extracted with 1 vol chloroform to remove any remaining phenol, and vortexed to mix layers. After centrifugation at room temperature for 5 min at 15000 × g, the aqueous phase containing the DNA was again removed, being careful not to collect or disturb interface. Next, 100 µL of 7.5 M (AAc) and 800 µL of absolute (100%) ethanol were added to each sample and mixed by inversion to precipitate DNA. Samples were placed at −20°C overnight. The next day, DNA was pelleted by centrifugation at 15000 × g for 15 min at room temperature. Supernatant was removed by pouring off or pipeting and the DNA pellet was rinsed in 70% ethanol and air dried until no moisture was apparent. After drying, DNA pellet was completely re-suspended in 200 µL of Solution B (Table 1) and stored at −20°C for further use. This extraction can be completed in 2 days, requiring 2–3 hours on day 1 and 1 hour on day 2. The maximum number of samples that can be processed simultaneously and still obtain quality DNA has not been determined. However, in our lab, one person has successfully processed up to 64 samples in a 2 day period.

Non-destructive DNA extraction protocol for dry peanut seed.
PCR Analysis of Transgenics . Polymerase chain reaction (PCR) amplification was performed to demonstrate the utility of DNA obtained from peanut seed using the above protocol. PCR was performed on DNA taken from dry F3 seed of transgenic peanut lines 487, 540, and 654. Primers used were constructed to amplify a 400 bp region of the hygromycin resistance gene (hph) contained in a plasmid construct that was used to generate the transgenic peanut lines (Chenault et al., 2002). Primer sequences were as follows: hph-forward 5′ TTTCTGATCGAAAAGT 3′ and hph-reverse 5′ AAGCTGCATCATCGAAATT 3′. PCR reactions took place in a total volume of 100 µL and contained 5 µL genomic DNA (25 ng/µL), 10 µL 10X PCR Buffer (500 mM KCl, 100 mM Tris-9.0 at 25°C, and 1% Triton® X-100 [Promega, Madison, WI]), 4 µL 25 mM MgCl2, 4 µL 10 mM dNTPs, 1 µL each primer (50 pmol/µL), 74.5 µL sterile water, and 0.5 µL Taq DNA polymerase (5 U/µL, Promega, Madison, WI). Amplification of all PCR products was performed using a PTC-100 thermal cycler (MJ research, Watertown, MA). Following an initial denaturation step for 2 min at 94°C, 35 cycles were carried out under the following conditions: 1 min at 94°C, 1 min at 55°C and 1 min at 72°C. A final extension was carried out at 72°C for 10 min. Negative controls (no template added) were included in all experiments.
SSR Analysis . Simple-sequence repeat (SSR) analysis was performed on genomic DNA taken from dry seed of the peanut cultivars and advanced breeding lines listed in the Plant Material section to further test the utility of DNA generated using this protocol. Sixteen different primer pairs were selected to examine polymorphism existing among the genotype test set (Ferguson et al., 2004). Amplification using each primer pair was carried out in a PTC-100 thermal cycler (MJ Research, Watertown, MA) under conditions optimized for each primer pair (Ferguson et al., 2004). Reaction components were as follows (20 µL total volume): 10 µl (2.5 ng/µl) genomic DNA, 2 µl 10X PCR Buffer (Qiagen, Valencia, CA), 2 µl 25 mM MgCl2, 1 µl each 10 µM Primers, 2 µl 2 mM dNTPs, 0.5 µl Hot Start Taq Polymerase (5 U/µl, Qiagen, Valencia, CA), 1.5 µl sterile water.
SCAR Analysis . Five crosses were made with the root-knot nematode resistant cultivar COAN (Simpson and Starr, 2001) and susceptible materials from the Florida peanut breeding program (Hull X COAN, COAN X Norden, COAN X UF98326, COAN X DP-2, and COAN X 920L19-8-1-1). F2 seed were produced from these crosses, collected, and DNA was extracted from them. The primers used for SCAR analysis were SCZ3-FO1 5′-CAGCACCGCAGCATAAAAAC-3′, and SCZ3-RO2 5′-CAGCACCGCACACATTCTGG-3′ (Garcia et al., 1996). PCR was carried out in a PTC-100 thermal cycler (MJ Research, Watertown, MA) as described in Garcia et al. (1996) under the following conditions: 94°C for 5 min, 30 cycles at 94°C for 1 min, 60°C for 1 min, 72°C for 2 min, and a final extension at 72°C for 3 min. A positive PCR reaction yielded a fragment of approximately 265 bp. Florunner was used as the negative control and COAN was used as the positive control.
Gel Electrophoresis . PCR products were separated by electrophoresis in a 1.0% agarose gel in 1 X Tris-Acetate-EDTA (TAE) buffer for 1.5 h at 100 V, and visualized by subsequent staining with ethidium bromide. The molecular marker used for PCR analysis was a 1 Kbp ladder (Invitrogen, Grand Island, NY). SSR fragments were separated on 3.5% MetaPhor agarose gel (Cambrex, Rockland, ME) in 1 X TAE for 6–7 h at 130 V and subsequently visualized by staining with ethidium bromide. The molecular marker used for SSR analysis was Trackit™ 25 bp DNA ladder (Invitrogen, Grand Island, NY).
Results and Discussion
Sample Quantity and Quality . The method described here can be used to consistently obtain high quality genomic DNA from peanut seed (Fig. 1). Yield from 200 samples averaged 38 ± 8 µg of DNA per extraction with an average A260/A280 ratio of 1.76 ± 0.3. With regards to the quantity of DNA extracted, this procedure is comparable to a mini-prep method developed for wheat seed (Hill-Ambroz et al., 2002) and to commercially available kits for use with plant material (Lickfeldt et al., 2002). In contrast to procedures developed for pre-germination screening of barley (von Post et al., 2003) and rice (Chunwongse et al., 1993) seed which produce a crude extract, this method provides clean peanut DNA which is suitable for downstream applications which require excellent DNA quality and quantity. Sampling did not appear to reduce germination or plantlet vigor. A germination rate of 95% was observed for seed that were germinated within 30 days of sampling [data not shown]. This rate was not significantly different from non-sampled seed of the same type. No testing of germination rate beyond this time period was performed.
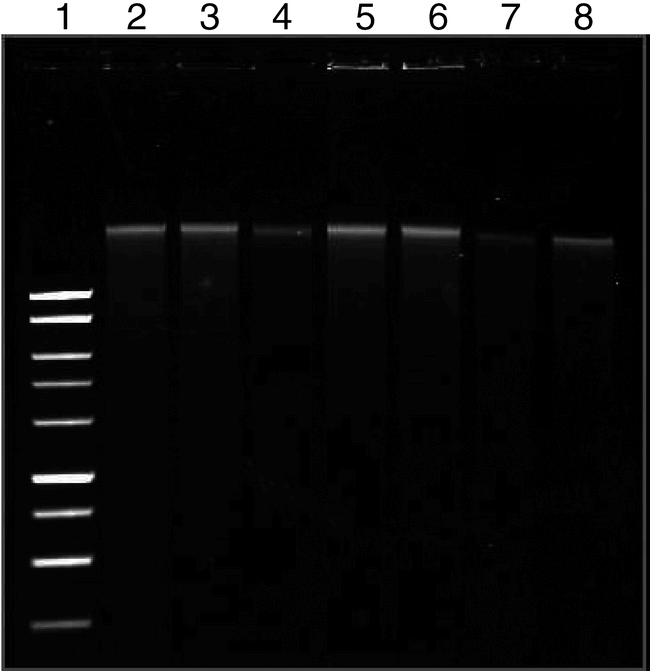
Genomic DNA extracted from peanut seed. Lane 1: 250 ng 1 kb ladder; Lanes 2–8: Independent peanut samples, 1 µl DNA from a 50 µl total volume (approx. 100 ng). The DNA was visualized on a 0.8% agarose, TAE gel and stained with 0.5 µg/ml ethidium bromide.
PCR Analysis of Transgenics . DNA taken from 100 R3 seed from each of three transgenic peanut lines previously shown to contain single copies of an hph marker gene/transgene construct (Chenault et al., 2002), were used to test the utility of this method for screening segregating populations. PCR analysis of the hph gene in the genomic DNA of seed from the transgenic peanut lines 487, 540, and 654 resulted in successful amplification demonstrated by the example in Fig. 2. The hph marker was observed in 72%, 79%, and 76% of the R3 seeds taken from lines 487, 540 and 654, respectively, confirming the 3∶1 ratio expected for this marker in an R3 population. Consistent amplification of the hph marker gene allowed the screening of the segregating population and thus the identification of those positive samples to be further analyzed and back-crossed. The use of this method saves time and resources compared to methods previously used to extract DNA from potentially transgenic peanut lines (Cheng et al., 1997; Magbanua et al., 2000; Chenault et al., 2002; Chenault and Payton, 2003; Livingstone et al., 2005) which involved isolating DNA from leaves.
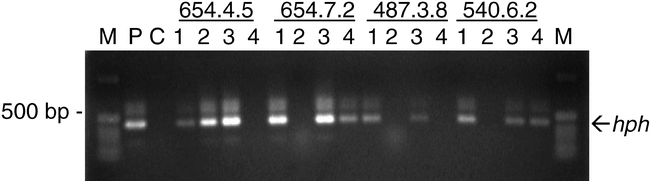
DNA fragments amplified from peanut genomic DNA taken from R3 seed of segregating transgenic lines 654, 487, and 540 using PCR primers hph forward and hph reverse. P indicates plasmid control; C indicates control amplifications in which DNA template was omitted. M indicates molecular weight standards.
SSR Analysis . DNA was extracted from the seed of 45 different genotypes encompassing all four peanut market types and was subjected to analysis by previously identified peanut SSR primers (Ferguson et al., 2004). In all, sixteen different primer pairs were used for analysis. Figs. 3a and 3b show examples of the banding patterns produced with the SSR primer pairs. Although not always polymorphic, successful amplification of SSR banding patterns was achieved using each primer pair/template combination, again demonstrating the utility of this DNA extraction method.

DNA fragments amplified from peanut genomic DNA taken from seed of cultivars and breeding lines representing all four market types using selected SSR primers. Lanes are labeled with genomic DNA type. (A) Banding patterns generated from genomic DNA using primer set 1 (B) Banding patterns generated from genomic DNA using primer sets 2 (upper lanes) and 3 (lower lanes). DNA samples in (B) are the same for both sets of lanes.
SCAR Analysis . DNA was extracted from 740 F2 seed from the five crosses listed in the Methods section along with seed from the positive control COAN and the negative control Florunner. The SCAR marker of 265 bp (Fig. 4) was observed for 193 F2 seeds (26%) and in seed of COAN. The cross COAN x Norden had the fewest seed with the SCAR marker (8.5%), and COAN X 920L19-8-1-1 had the most (59%). Presence of the SCAR marker was reproducible and a reliable indicator of field resistance to the root-knot nematode (> 98% correlation). An advantage of using MAS in this circumstance is the opportunity to screen peanut genotypes without relying on inoculation tests with the nematode, which is cumbersome and time-consuming. Additionally, MAS on the seed allowed putatively resistant individuals to be selected prior to planting which saves time and decreases the cost associated with planting and maintenance, by reducing the number of field plantings to those plants actually carrying the resistance gene in subsequent field trials. Peanut seed scored as positive for the SCAR marker have been successfully used in the Florida breeding program to quickly advance the development of root-knot nematode resistant lines suitable to conditions in the southeastern US.
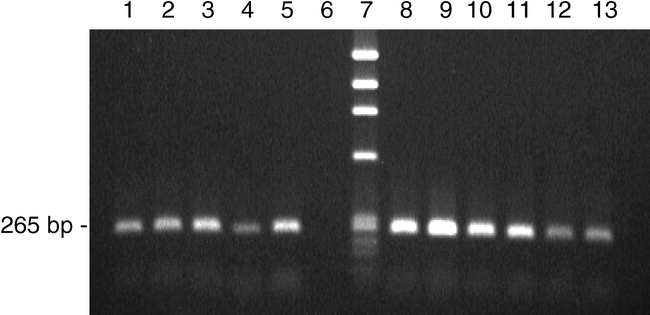
SCAR marker from peanut genomic DNA taken from seed of F2 populations segregating for nematode resistance. Lane 1, positive control COAN; Lanes 2 and 3, Hull x COAN; Lane 4, COAN x Norden; Lane 5, COAN x UF98326; Lane 6, negative control Florunner; Lane 7, Phi-X size marker; Lanes 8 and 9, COAN x DP-2; Lanes 10 and 11, COAN X 920L19-8-1-1; Lanes 12 and 13, COAN.
Conclusion
A method has been developed to successfully extract DNA from peanut seed in a non-destructive manner. DNA extracted using this method has been shown to be of excellent quality and useful for many PCR-based molecular techniques. Investigators can use this DNA extraction method to subject large populations of peanut seed to PCR-based selection and then germinate only the selected genotypes, therefore significantly enhancing the efficiency of molecular breeding and/or screening for transgenes among segregating populations.
Acknowledgements
The authors would like to express their appreciation to Lisa Myers for technical support. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. Portions of this research were funded by the Florida Agricultural Experiment Station.
Literature Cited
Bland J. M. and Lax A. R. 2000 Isolation and characterization of a peanut maturity-associated protein. J. Agric.Food Chem 48 : 3275 – 3279 .
Bolton M. D. , Nelson B. D. , Sparks R. B. , and Santoso A. 2005 Methods for extraction and amplification of DNA soybean seed. Seed Technol 27 : 89 – 94 .
Burrow M. D. , Simpson C. E. , Paterson A. H. , and Starr J. L. 1996 Identification of peanut (Arachis hypogaea L.) RAPD markers diagnostic of root-knot nematode (Meloidogyne arenaria (Neal) Chitwood) resistance. Mol. Breed 2 : 369 – 379 .
Charcosset A. and Moreau L. 2004 Use of molecular markers for the development of new cultivars and the evaluation of genetic diversity. Euphytica 137 : 81 – 94 .
Chenault K. D. , Burns J. A. , Melouk H. A. , and Payton M. E. 2002 Hydrolase activity in transgenic peanut. Peanut Sci 29 : 89 – 95 .
Chenault K. D. and Payton M. E. 2003 Genetic transformation of a runner-type peanut with the nucleocapsid gene of tomato spotted wilt virus. Peanut Sci 30 : 112 – 115 .
Cheng M. , Jarret R. L. , Li Z. , and Demski J. W. 1997 Expression and inheritance of foreign genes in transgenic peanut plants generated by Agrobacterium-mediated transformation. Plant Cell Rep 16 : 541 – 544 .
Choi K. , Burrow M. D. , Church G. , Burrow G. , Paterson A. H. , Simpson C. E. , and Starr J. L. 1999 Genetics and mechanism of resistance to Meloidgyne arenaria in peanut germplasm. J. Nematol 31 : 283 – 290 .
Chunwongse J. , Martin G. B. , and Tanksley S. D. 1993 Pre-germination genotypic screening using PCR amplification of half-seeds. Theor. Appl. Genet 86 : 694 – 698 .
Collard B. C. Y. , Jahufer M. Z. Z. , Brouwer J. B. , and Pang E. C. K. 2005 An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts. Euphytica 142 : 169 – 196 .
Demeke T. and Adams R. P. 1992 The effect of plant polysaccharides and buffer additives of PCR. BioTechniques 12 : 332 – 334 .
Ferguson M. E. , Burrow M. D. , Schulze S. R. , Bramel P. J. , Paterson A. H. , Kresovich S. , and Mitchell S. 2004 Microsatellite identification and characterization in peanut (A. hypogaea L.). Theor. Appl. Genet 108 : 1064 – 1070 .
Garcia G. M. , Stalker H. T. , Shroeder E. , and Kochert G. 1996 Identification of RAPD, SCAR, and RFLP markers tightly linked to nematode resistance genes introgressed from Arachis cardenasii into Arachis hypogaea. Genome 39 : 836 – 845 .
Herselman L. , Thwaites R. , Kimmins F. M. , Courtois B. , van der Merwe P. J. A. , and Seal S. E. 2004 Identification and mapping of AFLP markers linked to peanut (Arachis hypogaea L.) resistance to the aphid vector of groundnut rosette disease. Theor. Appl. Genet 109 : 1426 – 1433 .
Hill-Ambroz K. L. , Brown-Guedira G. L. , and Fellers J. P. 2002 Modified rapid DNA extraction protocol for high throughput microsatellite analysis in wheat. Crop Sci 42 : 2088 – 2091 .
Hird H. , Lloyd J. , Goodier R. , Brown J. , and Reece P. 2003 Detection of peanut using real-time polymerase chain reaction. Eur. Food Res. and Technol 217 : 265 – 268 .
Lickfeldt D. W. , Hofmann N. E. , Jones J. D. , Hamblin A. M. , and Voigt T. B. 2002 Comparing three DNA extraction procedures for cost, efficiency, and DNA yield. Hort Sci 37 : 822 – 825 .
Livingstone D. M. , Hampton J. L. , Phipps P. M. , and Grabau E. A. 2005 Enhancing resistance to Sclerotinia minor in peanut by expressing a barley oxidase gene. Plant Phys 137 : 1354 – 1362 .
Luo M. , Dang P. , Bausher M. G. , Holbrook C. C. , Lee R. D. , Lynch R. E. , and Guo B. Z. 2005 Identification of transcripts involved in resistance responses to leaf spot disease caused by Cercosporidium personatum in peanut (Arachis hypogaea). Phytopath 95 : 381 – 387 .
Magbanua Z. V. , Wilde H. D. , Roberts J. K. , Chowdhury K. , Abad J. , Moyer J. W. , Wetzstein H. Y. , and Parrott W. A. 2000 Field resistance to tomato spotted wilt virus in transgenic peanut (Arachis hypogaea L.) expressing an antisense nucleocapsid gene sequence. Mol. Breed 6 : 227 – 236 .
Melouk H. A. , Akem C. N. , and Brown C. 1992 A detached shoot technique to evaluate the reaction of peanut genotypes to Sclerotinia minor. Peanut Sci 19 : 58 – 62 .
Mohan M. , Nair S. , Bhagwat A. , Krishna T. G. , and Yano M. 1997 Genome mapping, molecular markers and marker-assisted selection in crop plants. Mol Breed 3 : 87 – 103 .
Paik-Ro O. G. , Smith R. L. , and Knauft D. A. 1992 Restriction fragment length polymorphism evaluation of six peanut species within the Arachis section. Theor. Appl. Genet 84 : 201 – 208 .
Pandey R. N. , Adams R. P. , and Flournoy L. E. 1996 Inhibition of random amplified polymorphic DNAs (RAPDs) by plant polysaccharides. Plant Mol. Biol. Reptr 14 : 17 – 22 .
Pataky J. K. , Black M. C. , Beute M. K. , and Wynne J. C. 1983 Comparative analysis of Cylindrocladium black rot resistance in peanut: greenhouse, microplot and field testing procedures. Phytopath 73 : 1615 – 1620 .
Sharma K. K. , Lavanya M. , and Anjaiah V. 2000 A method for isolation and purification of peanut genomic DNA suitable for analytical applications. Plant Mol. Biol. Reptr 18 : 393a – 393h .
Simpson C. E. and Starr J. L. 2001 Registration of ‘COAN’ peanut. Crop Sci 41 : 918 .
Sweeney P. M. and Danneberger T. K. 1997 RAPD markers for perennial ryegrass DNA extracted from seeds. Hort Sci 32 : 1212 – 1215 .
Sweeney P. , Golembiewski R. , and Danneberger K. 1996 Random amplified polymorphic DNA analysis of dry turfgrass seed. Hort.Sci 31 : 400 – 401 .
von Post R. , von Post L. , Dayteg C. , Nilsson M. , Forster B. P. , and Tuvesson S. 2003 A high-throughput DNA extraction method for barley seed. Euphytica 130 : 255 – 260 .
Notes
- 1USDA-ARS, Wheat, Peanut and Other Field Crops Research Unit, 1301 N. Western, Stillwater, OK 74075. [^]
- 2Agronomy Department, Plant Molecular and Cellular Biology Program, and The Genetics Institute, University of Florida, Gainesville, FL 32611. [^]
- 3Agronomy Department, Cancer/Genetics Research Complex, University of Florida, Gainesville, FL 32611. [^] *Corresponding Author: (kelly.chenault@usda.ars.gov)
Author Affiliations